The following side project, conducted during the period between my thesis defence and the submission of corrections, has been published as a review article titled Locked in Structure: Sestrin and GATOR—A Billion-Year Marriage, available at:
During the preparation of my thesis, I sought to incorporate a bioinformatics component into the Introduction chapter. I constructed a phylogenetic tree of Sestrin proteins using ClustalOmega and BLAST. The resulting tree was used for a figure in my thesis (Figure 1), displaying 805 Sestrins and their homologs, highlighting different groups of organisms in which they were found.
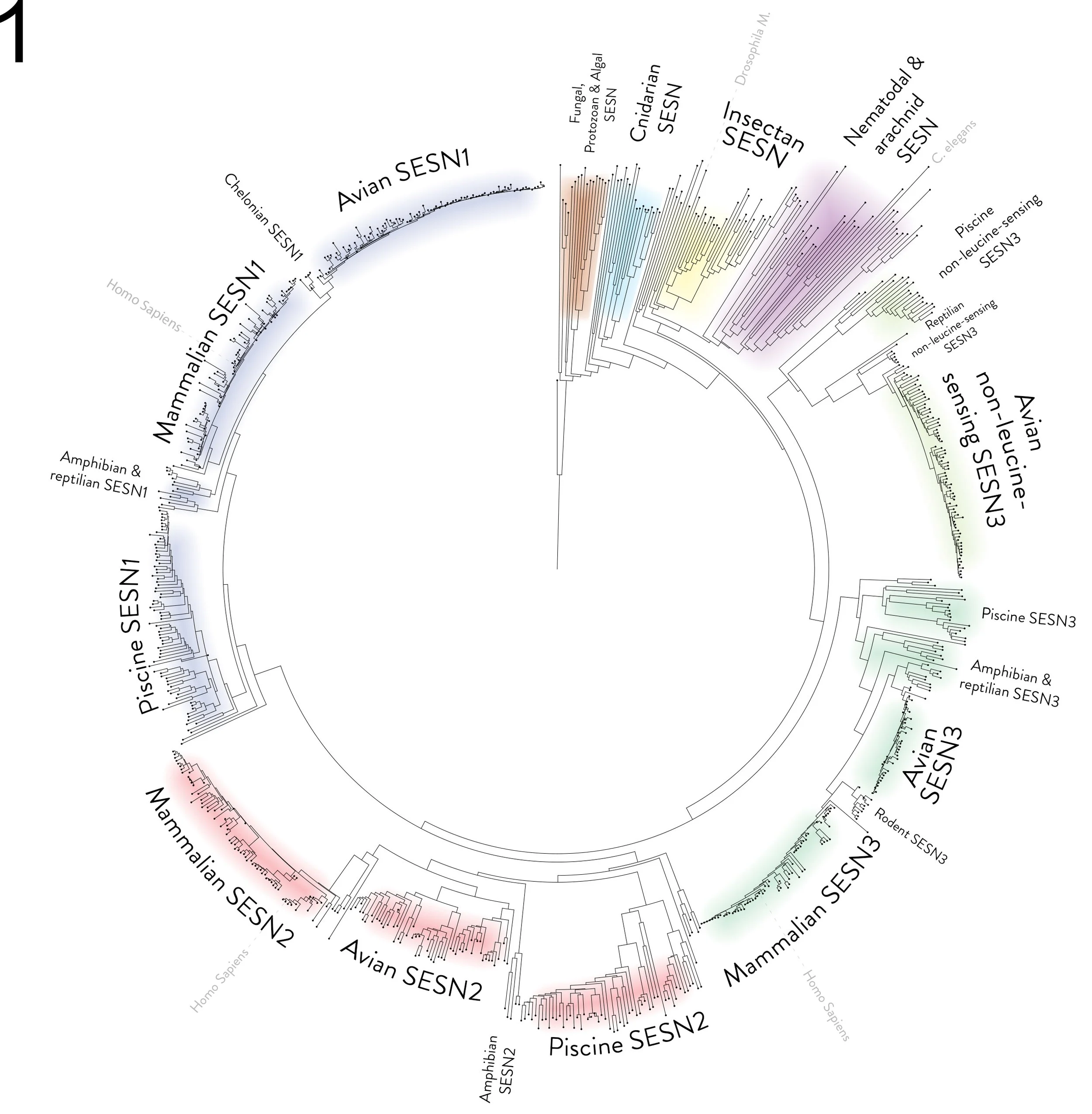
Figure 1 - Phylogenetic tree based on Sestrin protein sequences
A circular phylogenetic tree based on protein alignments of Sestrin. The tree displays 805 Sestrin and Sestrin-like proteins as nodes at the tip of a leaf. Protein BLAST was used to select species with homologous proteins to the C. Elegans Sestrin sequence. One species of every genus was selected manually via BLAST's taxonomic display, and FASTA sequences were retrieved for each protein. A multiple sequence alignment was performed using EMBL-EBI Clustal Omega. The programme was used to export the alignment as a Newick format phylogenetic tree. The file was imported into the Interactive Tree Of Life (iTOL) programme, and the tool was used to root the tree on the sequence of Naegleria gruberi, and some visual aspects of the tree were adjusted. The figure of the circular tree was exported as an image file. The tree was then annotated and coloured manually using Adobe Photoshop. As this is a protein-aligned phylogenetic tree, the distances on the tree are arbitrary. The distance from a node to the tip of a leaf represents the difference in the sequence from a previously assumed ancestor protein. E.g. all avian SESN1 seem to be highly similar, hence very close to the same line.
To further explore the evolutionary origins of Sestrins, we expanded the dataset of proteins. Using BLAST, we selected the longest isoform of Sestrin from each identified genus, resulting in an alignment of 1006 proteins (covering SESN1, SESN2, and SESN3) across 575 genera and 587 species. This analysis spanned metazoan species from humans to Caenorhabditis elegans and was dubbed the ‘metazoan’ alignment. Additionally, in a separate alignment, we extended our analysis beyond metazoan genera. We used the putative Sestrin from Naegleria gruberi as the BLAST query and identified 213 homologous proteins across 131 genera. This was dubbed the ‘non-metazoan’ alignment.
The alignments were analysed using Jalview, and conservation percentages were calculated for each residue of the Sestrin protein. Motif discovery was then performed with the MEME (Multiple Em for Motif Elicitation) suite. In combination, the identified motifs were examined for conservation and the functional relevance of these regions was discussed, including a comparison between metazoan and non-metazoan Sestrins (Figure 2).
We also reviewed the published site-directed mutagenesis studies that were previously performed on Sestrins and attempted to analyse them in light of our conservation analysis. This analysis revealed that several residues essential for leucine sensing are conserved only in metazoans, suggesting that this function likely evolved within the metazoan lineage (Figure 2B, C). In contrast, the residue associated with antioxidant activity, the catalytic cysteine (C125), was conserved in both metazoans and non-metazoans, indicating an earlier evolutionary origin (Figure 2A). Interestingly, residues implicated in GATOR2 binding (e.g. the DDYDY and WSLAEL motifs) were also partially conserved in non-metazoans, often by chemically similar residues, suggesting that the ability to interact with GATOR2 or GATOR2-like proteins may have emerged prior to the highly conserved metazoan version of Sestrin (Figure 2D, E).
The full alignment results and motif analyses are available as Supplementary Materials in the article Locked in Structure: Sestrin and GATOR—A Billion-Year Marriage.

Figure 2 - MEME and conservation analysis
Conservation of Sestrin motifs that are responsible for the unique functions of Sestrin. A Motif MAARQCSYL that is responsible for the antioxidative function. B, C Motifs that are implicated in leucine sensing. D Motif WSLAEL and E motif DDYDY are responsible for binding to GATOR2.
The readout of the MEME suite motif discovery tool is displayed in a histogram of letters. An Excel representation of our alignment analysis is presented. The rows represent the following: row 1—position on SESN2; row 2—residue on SESN2; row 3—alignment consensus at this position; row 4—the % of entries showing consensus; row 5—the number of entries showing consensus (Metazoan N = 1006, Non-metazoan N = 213). The position on the SESN2 structure (PDB ID: 5DJ4) is shown on the right.
To further investigate the SESN2-GATOR2 relationship, we performed a conservation analysis of the GATOR2 subunits WDR24 and SEH1L. It was previously demonstrated that SESN2 binds somewhere on the β-propeller domains of these proteins. By mapping conserved residues within these β-propeller domains and examining known GATOR2-binding sites on SESN2, we proposed a putative binding geometry (Figure 3).
Notably, a recent Nature study [1] displayed the structure of bound SESN2 and GATOR2, revealing that several highly conserved arginine residues in WDR24 (>99% conservation) directly participate in SESN2 binding. This highlights the usefulness of conservation analysis as a predictive tool to guide hypotheses and experimental design prior to wet-lab work.

Figure 3 - Sestrins bind somewhere along the WDR24-SEH1L β-propellers
The figure displays different representations of the WDR24-SEH1L β-propeller domains. A (a) The location of the WDR24-SEH1L β-propellers on the GATOR2 structure (PDB ID: 7UHY). (b) The cartoon representation of WDR24-SEH1L β-propellers. Green—WDR24; Red—SEH1L. (c) A top-down view of the blade donation from WDR24 to SEH1L, corresponding to (b), rotated by 90 degrees. The rest of the WDR24 structure is faded for visual clarity. B The results of our alignment analysis are visualised as shades of green on the WDR24-SEH1L arrangement. The legend for the shades of colour is displayed in the corner of the figure. (a) The cartoon representation of WDR24-SEH1L β-propellers and their conserved residues. (b) Surface representation of residues from the image in (a). (c) A view of the back of the arrangement, where (b) has been rotated along the y-axis by 180 degrees. C The hypothetical arrangement of the SESN2-GATOR2 binding. (a) A side view of the WDR24-SEH1L arrangement; the view above in B has been rotated by 90 degrees to the right along the y-axis. (b) The cartoon representation of the SESN2 face that binds GATOR2, with important sites annotated.
To trace the structural ancestry of SESN2, we employed AlphaFold and PyMOL to identify proteins with similar tertiary structures. Tertiary structure is often conserved despite substitutions in the primary sequence with chemically similar residues, making structural similarity a powerful indicator of evolutionary relationships. The AhpD from S. pneumoniae adopts a dimeric quaternary structure. Using PyMOL, we generated an artificial fused dimer by bonding two AhpD monomers using PyMOL’s bond command. We aligned the resulting fused dimer of AhpD to the human SESN2, and remarkably, this fused AhpD dimer overlapped well with human SESN2 (RMSD = 3.3 Å).
We then searched the AlphaFold Clusters database (Steinegger Lab) with the fused AhpD dimer as the query structure. This revealed several bacterial proteins that were structurally analogous to a fused AhpD dimer. A subset of these structures was examined, and their potential evolutionary relevance to Sestrins was discussed (Figure 4).

Figure 4 - The ancestry of Sestrins
The figure displays the bacterial structural analogues of Sestrins. The hypothetical evolutionary route is illustrated from left to right, beginning with the monomeric spAhpD and ending with the human SESN2. The structures of the bacterial proteins were aligned to SESN2 using PYMOL, and Mol* Viewer was used to generate cartoon representations at the same angle for visual clarity. PDB IDs are annotated, and for unresolved structures, the AlphaFold predictions were used. The RMSD values of the structural alignment to SESN2 are as follows: spAhpD—RMSD 3.353093 over 152 residues; Coma_aqua—RMSD 2.967612 over 144 residues; Cory_urei—RMSD 3.812889 over 152 residues; Candid_Roku—RMSD 4.020528 over 144 residues; YciW—RMSD 6.050121 over 144 residues; SESN_Naeg—RMSD 3.472114 over 184 residues. Red—SESN-A; Green—SESN-C.
1. Valenstein ML, Wranik M, Lalgudi PV, Linde-Garelli KY, Choi Y, Chivukula RR, Sabatini DM, Rogala KB: Structural basis for the dynamic regulation of mTORC1 by amino acids. Nature 2025, 646:493-500.